Your partner in science for end-to-end CRDMO solutions
Jubilant Biosys' Ligand-Based Drug Discovery Services
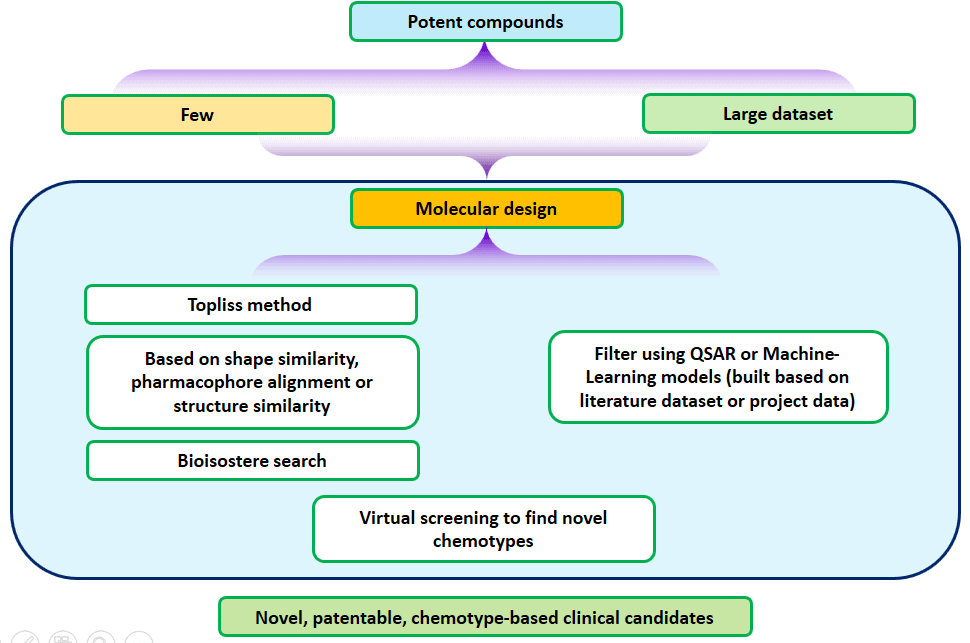
In the field of drug discovery, very often, the target structure will not be available, but a few small molecules might be known to be modulating the target. In such a situation, the small molecules are used as the basis for discovery efforts. Ligand-based drug discovery (LBDD) refers to drug discovery efforts in absence of any target structures and in presence of chemical structures known to modulate the target.

Ligand-based drug discovery
Ligand-based drug discovery starts with either a single compound or a set of compounds known to be potent against a target and based on the knowledge of structure-activity relationships (SAR), potency and other important properties are improved by designing appropriate analogs. Designing can be accomplished by Topliss method or simple analog design based on structural similarity or properties. Often, computational tools such as pharmacophore models or shape of the compounds can be useful for design purposes. Once a dataset of considerable size becomes available, with a good range of potency, a Quantitative Structure-Activity Relationships (QSAR) models can be attempted and used if the models are robust enough for prediction purposes. Similarly, if the target is well known with a lot of compounds already known in public literature or public databases, then machine-learning based models can also be attempted. If the machine-learning models are robust enough, they can be used for filtering design ideas or virtual screening or scaffold-hopping hits. Jubilant has been successful in delivering clinical candidate compounds for several targets for which the target structure was not known at the time. The computational chemistry team, along with the medicinal chemistry team, collaborates quite closely, to run the projects requiring LBDD efforts.
Jubilant Biosys employs Schrodinger software suite as well as Cresset’s scaffold-hopping tool, Spark, for driving ligand-based drug discovery projects. The computational chemistry group of Jubilant Biosys also employs many machine-learning (ML) algorithms to build ML-based models, both regression and classification models, to assist LBDD efforts.
If the target structure is not known, but structures of its closest homologues are known, then a homology-based model can be built using the experimental coordinates of the structure of the closest homologue. If the homology model is good enough, then a structure-based design strategy can be employed.
Jubilant Biosys provides a wide variety of specialized support for drug discovery services.
If you are looking for quality solutions incorporated with the best practices in the drug discovery domain, feel free to contact us.