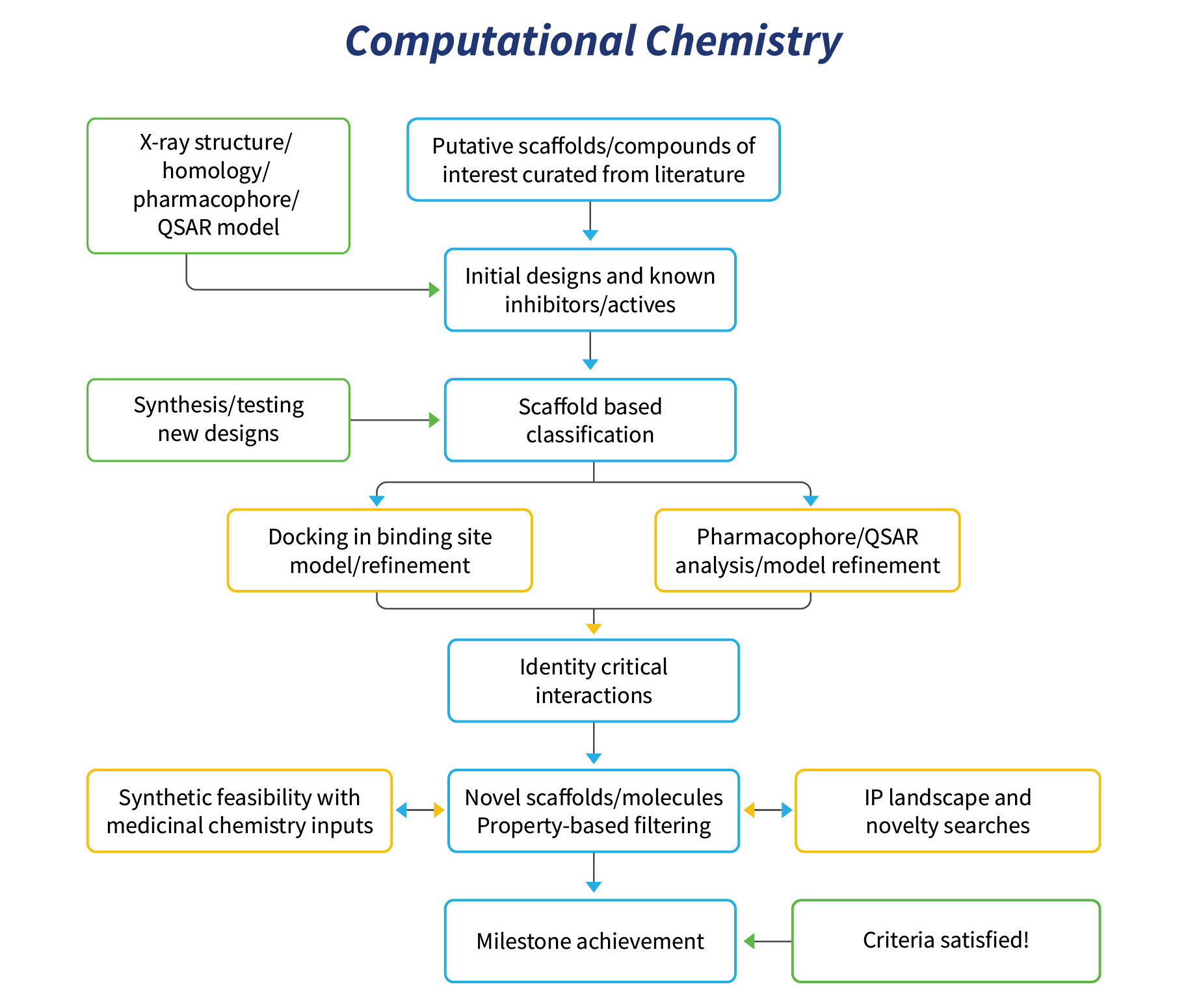
The team providing these computational chemistry services extensively utilizes in-house expertise in software development, curation and structural biology for driving molecular design.
Achievements
Molecular Design Collaborations
Our team of computational chemists empowers drug discovery scientists to deploy ready-to-use models and make faster and wiser decisions.
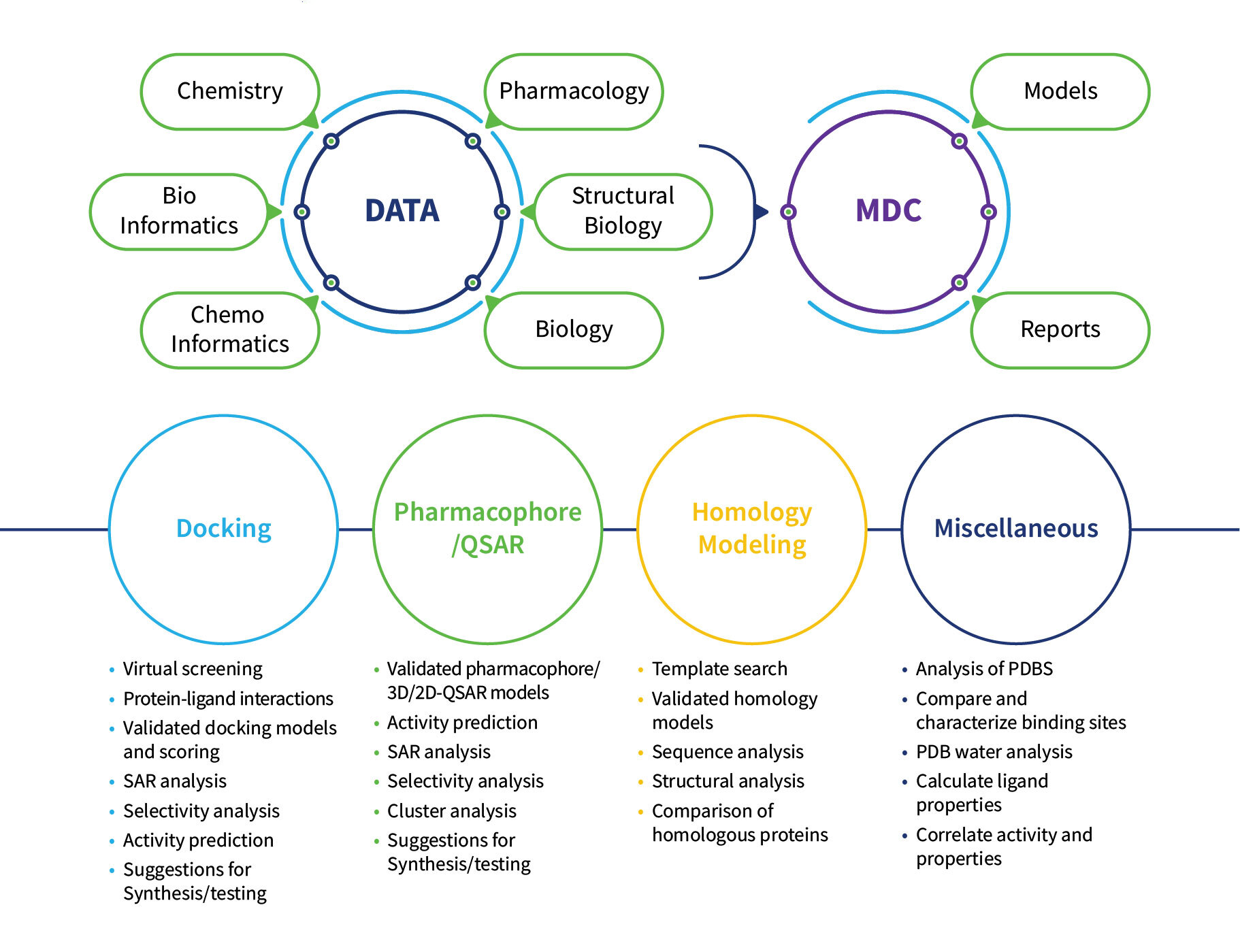
The Tools Used for Computational Drug Discovery
The People Providing Expertise in a Range of Computational Chemistry Solutions